eCTD (Electronic Common Technical Document), is a standard format and interface for electronic delivery of documentation submitted to Medicines Agency for the purposes of registration of medicines or variations. eCTD structure is based on CTD format (Common Technical Document).
The intensive preparations are underway for the implementation of electronic filing and processing applications (eCTD) in practice. Launch of this process is by the end of 2011. For the success of the eCTD project is inavitable that applicants are familiar with the requirements to fill an application and its identification.
Up to 10th June 2011, 37 applications submitted to SIDC in eCTD format have been tested. Only 3 (~8%) applications fully meet the validation criteria of eCTD version 3.1.
For More Information Please click here: SIDC(Slovakian Agency) Ongoing eSubmissions Issues
30 June 2011
Apex Regulatory Blog: Document Management
The importance of effective document management can’t be overstated in founding and maintaining an efficient, well ordered and audit ready Regulatory department. DMSs such as SCORE or Documentum’ FirstDoc are powerful tools which primarily act as a repository for your company’s documents. But they can do so much more than this. They are designed to: interact with publishing software, enable document workflows, archive documents, provide audit trails…there are too many features to list here!
So, what do I mean by ‘effective document management’? Well, from my experience working in a number of systems and seeing first hand the nightmares that substandard document management can cause.
For More Information Please click here: Apex Regulatory Blog: Document Management
So, what do I mean by ‘effective document management’? Well, from my experience working in a number of systems and seeing first hand the nightmares that substandard document management can cause.
For More Information Please click here: Apex Regulatory Blog: Document Management
JAZMP (Slovenian Agency): INSTRUCTIONS FOR THE SURRENDER OF APPLICATIONS AND VALIDATION CRITERIA IN ECTD and NeeS FORM
The purpose of this guide is to fully define the requirements of the Public Agency for Medicinal Products and medicinke devices (hereinafter JAZMP) technical validation for successful applications in the format e-CTD and NeeS.
Scope
Guidance should be used by applicants who apply on JAZMP role in electronic format or e-CTD NeeS.
E-CTD format or NeeS investors can apply for the following types of applications:
1st Authorisation Holder,
2nd Expansion of an existing permit holder,
3rd Renewal of Marketing Authorisation
4th Changes to the terms of marketing,
5th Periodic Safety Update Report (PSUR);
JAZMP accepts such types of applications for MRP, DCP, NP and CP procedures in human medicine for procedures for medicinal products for veterinary use and accept electronic applications in VNeeS format.
For More Information Please click here: JAZMP (Slovenian Agency) eCTD & Nee'S Updates
Scope
Guidance should be used by applicants who apply on JAZMP role in electronic format or e-CTD NeeS.
E-CTD format or NeeS investors can apply for the following types of applications:
1st Authorisation Holder,
2nd Expansion of an existing permit holder,
3rd Renewal of Marketing Authorisation
4th Changes to the terms of marketing,
5th Periodic Safety Update Report (PSUR);
JAZMP accepts such types of applications for MRP, DCP, NP and CP procedures in human medicine for procedures for medicinal products for veterinary use and accept electronic applications in VNeeS format.
For More Information Please click here: JAZMP (Slovenian Agency) eCTD & Nee'S Updates
GlobalSubmit: Clinical Study Reports to STFs
What are STFs, how are STFs used, and why are STFs good?
Study Tagging Files, or STFs, organize study information into meaningful, standardized headings, not provided by the eCTD DTD, which allows reviewers to quickly understand what has been submitted and what has not. STFs provide consistency over the lifecycle of the regulatory application.
An STF should be provided with the submission of any file, or group of files belonging to a study in Modules 4 and 5. STFs are required by the United States, are not required in Europe and Canada, and are not allowed in Japan.
To understand STFs sufficiently you need to understand the index.xml.
For more information click here: GlobalSubmit STF
Study Tagging Files, or STFs, organize study information into meaningful, standardized headings, not provided by the eCTD DTD, which allows reviewers to quickly understand what has been submitted and what has not. STFs provide consistency over the lifecycle of the regulatory application.
An STF should be provided with the submission of any file, or group of files belonging to a study in Modules 4 and 5. STFs are required by the United States, are not required in Europe and Canada, and are not allowed in Japan.
To understand STFs sufficiently you need to understand the index.xml.
For more information click here: GlobalSubmit STF
27 June 2011
Beyond eCTD: The Regulated Product Submissions Standard by Joel Finkle
Beyond eCTD: The Regulated Product Submissions Standard by Joel Finkle - Senior Strategist, Regulatory Informatics, CSC.
Please click here to find the Article: Beyond eCTD: The Regulated Product Submissions Standard by Joel Finkle
Please click here to find the Article: Beyond eCTD: The Regulated Product Submissions Standard by Joel Finkle
25 June 2011
Paperless Submission - Current Situation in the EU By Rob de Haan & Alaistair Nixon
Presentaion of Paperless Submission - Current Situation in the EU from Rob de Haan of MEB and Alaistair Nixon of GSK Regulatory Publishing. Including eCTD and NeeS Submissions.
Please click here to find the presentation: Paperless Submission - Current Situation in the EU
Please click here to find the presentation: Paperless Submission - Current Situation in the EU
Ask Cato:Is FDA Issuing More Refuse-to-File Letters?
The June 2011 issue of Nature Reviews: Drug Discovery (volume 10, page 403) includes an interesting news brief regarding the apparent increase in Refuse-to-File (RTF) letters being issued by FDA in recent years.
In a review of publicly available RTF letters, researchers at Leerink Swann found that 15 RTF letters were issued from 1998 to 2009 and that there have already been 13 issued since 2009. Among the reasons for given for several RTFs in the last two years.
For More Information Please Click Here: Ask Cato:Is FDA Issuing More Refuse-to-File Letters?
In a review of publicly available RTF letters, researchers at Leerink Swann found that 15 RTF letters were issued from 1998 to 2009 and that there have already been 13 issued since 2009. Among the reasons for given for several RTFs in the last two years.
For More Information Please Click Here: Ask Cato:Is FDA Issuing More Refuse-to-File Letters?
Webinar: How to Avoid the Dreaded Refuse-to-File, Plus 2012 eCTD Mandate from Congress - Sept. 6, 2011, 2-3 PM EDT - By Antoinette Azevedo
One of our most praised presentations, this Webinar by renowned eCTD expert Antoinette Azevedo will provide an overview of best practices to enable a sponsor to prepare a compliant eCTD for regulatory authority review. So you can avoid a costly Refuse-to-File (RTF) by FDA.
6 Things You will Learn:
Top issues regulatory authorities have with eCTD and how to avoid them.
Preparing submission-ready source documents and datasets for submission in eCTD.
Whether to purchase an eCTD publishing system or to outsource.
The role of an electronic document management system (EDMS) in an eCTD publishing solution?
How to prepare for the technical challenges of eCTD.
How to interact with regulatory authorities to assure your eCTD submission is accepted for review.
For More Information Please Click Here How to Avoid the Dreaded Refuse-to-File, Plus 2012 eCTD Mandate from Congress
6 Things You will Learn:
Top issues regulatory authorities have with eCTD and how to avoid them.
Preparing submission-ready source documents and datasets for submission in eCTD.
Whether to purchase an eCTD publishing system or to outsource.
The role of an electronic document management system (EDMS) in an eCTD publishing solution?
How to prepare for the technical challenges of eCTD.
How to interact with regulatory authorities to assure your eCTD submission is accepted for review.
For More Information Please Click Here How to Avoid the Dreaded Refuse-to-File, Plus 2012 eCTD Mandate from Congress
22 June 2011
CDISC Releases Final v3.0 Standard for Exchange of Nonclinical Data (SEND) Implementation Guide
The CDISC SEND Team is pleased to announce the release of the SEND Implementation Guide (IG) Version 3.0.
The SENDIG is intended to guide the organization, structure, and format of standard nonclinical tabulation datasets for interchange between organizations such as sponsors and CROs and for submission to the US Food and Drug Administration (FDA).
Version 3.0 of the SENDIG in now the production version and is designed to support single-dose general toxicology, repeat-dose general toxicology, and carcinogenicity studies. The SENDIG is based upon and should be used in close concert with Version 1.2 of the CDISC Study Tabulation Model (SDTM).
For More Information Click here: CDISC Nonclinical Data
The SENDIG is intended to guide the organization, structure, and format of standard nonclinical tabulation datasets for interchange between organizations such as sponsors and CROs and for submission to the US Food and Drug Administration (FDA).
Version 3.0 of the SENDIG in now the production version and is designed to support single-dose general toxicology, repeat-dose general toxicology, and carcinogenicity studies. The SENDIG is based upon and should be used in close concert with Version 1.2 of the CDISC Study Tabulation Model (SDTM).
For More Information Click here: CDISC Nonclinical Data
21 June 2011
Regulatory Analysis: Electronic Regulatory Submissions - Barbara Jentges, PhACT
With electronic submissions (e-submissions) following the internationally standardized Common Technical Document (eCTD) format, the regulatory submission has changed from paper to digital.
The specification for the eCTD is based on “Extensible Markup Language” (XML) technology and lists the criteria that make an e-submission technically valid. It focuses on “the ability to transfer the registration application electronically from industry to a regulatory authority.” The data and document files that form part of a submission are mostly provided in a portable document format (PDF) and are embedded into the XML backbone of the eCTD.
The change from paper to e-submissions bears some challenges: The implementation of suitable information and communication technology (ICT), the adaption of related business processes, as well as working practices, and particularly, the professional use of ICT play key roles for a successful e-submission project.
As brought to the point in Europe’s Digital Competitiveness Report 2010: “Investment in ICT is not sufficient if not accompanied by the reorganization of internal processes. ICT applications that help automatic business processes can be an important source of efficiency gains when accompanied by innovative working practices and the appropriate skills.
In this context, particularly in view of Europe’s digital agenda for a flourishing digital economy by 2020, it is worth exploring whether current practices can allow the efficient handling of an eCTD submission project.
“eCTD-compliant” Navigable Files
One major advantage of the eCTD is its ability to allow navigation through a complete electronic submission. However, the eCTD can only be navigated provided that each file is navigable itself and provided referenced files are hyperlinked with each other.
In order to become “navigable,” the PDF files need to fulfill specific formal requirements (e.g., document granularity, specific file names) and need specific properties which allow navigation, like bookmarks, intra- and inter-text hyperlinks (see Figure 1). The properties of a PDF file are specified in a U.S. FDA guidance and in a number of regional eCTD-related guidance documents.

If the author or writer of a document does not consider these requirements, a file needs to go through numerous time-consuming formatting steps until it is ready for a submission in eCTD format (for examples, see Figure 2). That happens mainly when the files are provided by external parties, e.g., Clinical Research Organizations (CROs) or medical writers.

The major efforts of time-consuming reformatting can be prevented by standardizing the document format. This can be achieved by either using harmonized templates and/or by setting up a style guide where aspects of document granularity, document formats, hyperlinking, etc., are specified. However, all parties/persons providing submission-relevant files need to be informed and trained, if necessary, in the document format standardization or as the U.S. FDA points out on its related website “Ask CROs in advance to provide reports in searchable PDF format compliant with the ICH M4 Granularity Annex and FDA PDF Specification.”
Even when working with standardized document format, interoperability problems may occur by using different operating systems and software standards (e.g., by using different Microsoft® Office software versions). Before exchanging any submission-relevant files with external parties, an interoperability check may help to identify and prevent the problems.
The need for “interoperability and standards” was identified as one of the seven actions in Europe’s Digital Agenda Communication initiated to make “proposals for actions that need to be taken urgently to get Europe on track for smart, sustainable and inclusive growth….”
Adapting Business Processes, Working Practices & Improving e-Skills
Working with electronic files rather than paper requires the adaption of eCTD-related business processes.
The eCTD submission project can only be efficient when the underlying business processes are accompanied by innovative e-working practices and the appropriate e-skills.
In practice, however, and as listed below, business processes and working practices sometimes run counter to an efficient eCTD submission project processing, especially in small and medium-sized enterprises (SMEs).
The following are common challenges an eCTD submission project can face, but it is certainly not an exhaustive list:
•Inadequate or even missing project management skills. There is a risk that the eCTD submission project could get out of control and be behind schedule.
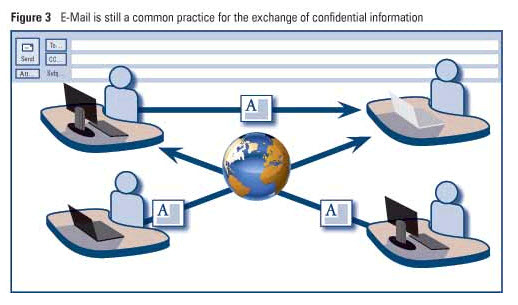
•Unnecessary multiplication of documents and bytes when large and confidential files are exchanged via email (see Figure 3). A secure, web-based virtual project place (like Microsoft® SharePoint®) is needed as a document repository that is accessible for all project members and enables the collaboration on documents and data (see Figure 4 ).
•A lack of e-skills, resulting in:
– missing interdisciplinary communication with ICT specialists regarding ICT-needs (either company-internal or service provider)
– suboptimal use of software (using the computer as a “typewriter” without being aware of useful software functionalities or the information about additional software to optimize specific working practices)
– Lack of knowledge about electronic document properties and how to change them (image-based versus text-based PDFs, protected PDFs, file size reduction, inherit zoom magnification, file size reduction, etc.)


In light of these challenges, the following questions arise:
Do regulatory affairs professionals bring along the required e-skills to adapt innovative e-working practices in order to manage a complex eCTD submission project? If not, what measures will become necessary?
One answer was given in last year’s European e-Skills Conference: “Organizations need to invest not only in infrastructure but in the higher level e-skills of their workforce…. The critical factor for future success…is the capacity for the competitive application of technologies.
In conclusion and in view of Europe’s digital agenda, the time has come for Good e-Submission Practice.
About the Author
Barbara is based in Switzerland. She is the Managing Director of PhACT GmbH; a company that provides advice and service in drug regulatory affairs, with a specialty in EU regulatory submissions including biotechnology. Barbara has more than 20 years of experience in regulatory affairs and previously worked with the Federal Institute for Drugs and Medicinal Devices (BfArM)-the German Health Authorities.
The specification for the eCTD is based on “Extensible Markup Language” (XML) technology and lists the criteria that make an e-submission technically valid. It focuses on “the ability to transfer the registration application electronically from industry to a regulatory authority.” The data and document files that form part of a submission are mostly provided in a portable document format (PDF) and are embedded into the XML backbone of the eCTD.
The change from paper to e-submissions bears some challenges: The implementation of suitable information and communication technology (ICT), the adaption of related business processes, as well as working practices, and particularly, the professional use of ICT play key roles for a successful e-submission project.
As brought to the point in Europe’s Digital Competitiveness Report 2010: “Investment in ICT is not sufficient if not accompanied by the reorganization of internal processes. ICT applications that help automatic business processes can be an important source of efficiency gains when accompanied by innovative working practices and the appropriate skills.
In this context, particularly in view of Europe’s digital agenda for a flourishing digital economy by 2020, it is worth exploring whether current practices can allow the efficient handling of an eCTD submission project.
“eCTD-compliant” Navigable Files
One major advantage of the eCTD is its ability to allow navigation through a complete electronic submission. However, the eCTD can only be navigated provided that each file is navigable itself and provided referenced files are hyperlinked with each other.
In order to become “navigable,” the PDF files need to fulfill specific formal requirements (e.g., document granularity, specific file names) and need specific properties which allow navigation, like bookmarks, intra- and inter-text hyperlinks (see Figure 1). The properties of a PDF file are specified in a U.S. FDA guidance and in a number of regional eCTD-related guidance documents.

If the author or writer of a document does not consider these requirements, a file needs to go through numerous time-consuming formatting steps until it is ready for a submission in eCTD format (for examples, see Figure 2). That happens mainly when the files are provided by external parties, e.g., Clinical Research Organizations (CROs) or medical writers.

The major efforts of time-consuming reformatting can be prevented by standardizing the document format. This can be achieved by either using harmonized templates and/or by setting up a style guide where aspects of document granularity, document formats, hyperlinking, etc., are specified. However, all parties/persons providing submission-relevant files need to be informed and trained, if necessary, in the document format standardization or as the U.S. FDA points out on its related website “Ask CROs in advance to provide reports in searchable PDF format compliant with the ICH M4 Granularity Annex and FDA PDF Specification.”
Even when working with standardized document format, interoperability problems may occur by using different operating systems and software standards (e.g., by using different Microsoft® Office software versions). Before exchanging any submission-relevant files with external parties, an interoperability check may help to identify and prevent the problems.
The need for “interoperability and standards” was identified as one of the seven actions in Europe’s Digital Agenda Communication initiated to make “proposals for actions that need to be taken urgently to get Europe on track for smart, sustainable and inclusive growth….”
Adapting Business Processes, Working Practices & Improving e-Skills
Working with electronic files rather than paper requires the adaption of eCTD-related business processes.
The eCTD submission project can only be efficient when the underlying business processes are accompanied by innovative e-working practices and the appropriate e-skills.
In practice, however, and as listed below, business processes and working practices sometimes run counter to an efficient eCTD submission project processing, especially in small and medium-sized enterprises (SMEs).
The following are common challenges an eCTD submission project can face, but it is certainly not an exhaustive list:
•Inadequate or even missing project management skills. There is a risk that the eCTD submission project could get out of control and be behind schedule.
•Unnecessary multiplication of documents and bytes when large and confidential files are exchanged via email (see Figure 3). A secure, web-based virtual project place (like Microsoft® SharePoint®) is needed as a document repository that is accessible for all project members and enables the collaboration on documents and data (see Figure 4 ).
•A lack of e-skills, resulting in:
– missing interdisciplinary communication with ICT specialists regarding ICT-needs (either company-internal or service provider)
– suboptimal use of software (using the computer as a “typewriter” without being aware of useful software functionalities or the information about additional software to optimize specific working practices)
– Lack of knowledge about electronic document properties and how to change them (image-based versus text-based PDFs, protected PDFs, file size reduction, inherit zoom magnification, file size reduction, etc.)

In light of these challenges, the following questions arise:
Do regulatory affairs professionals bring along the required e-skills to adapt innovative e-working practices in order to manage a complex eCTD submission project? If not, what measures will become necessary?
One answer was given in last year’s European e-Skills Conference: “Organizations need to invest not only in infrastructure but in the higher level e-skills of their workforce…. The critical factor for future success…is the capacity for the competitive application of technologies.
In conclusion and in view of Europe’s digital agenda, the time has come for Good e-Submission Practice.
About the Author
Barbara is based in Switzerland. She is the Managing Director of PhACT GmbH; a company that provides advice and service in drug regulatory affairs, with a specialty in EU regulatory submissions including biotechnology. Barbara has more than 20 years of experience in regulatory affairs and previously worked with the Federal Institute for Drugs and Medicinal Devices (BfArM)-the German Health Authorities.
16 June 2011
eCTD Tips: Microsoft Office 2010 and PDF Creation Problems
In a previous post, I discussed a minor issue that we encountered when my company recently upgraded from Microsoft Office 2003 to Microsoft Office 2010. In this post, I’ll describe a more serious problem caused by the update and the (relatively) easy solution we finally discovered.
The Problem
Prior to the upgrade, we used the “Create a PDF” button on the Acrobat toolbar in Word to create PDFs for submissions. As you know, PDFs created using this button will include functional bookmarks based upon the heading styles used, a fully hyperlinked table of contents, and functional internal hyperlinks within the text of the document (if they were created by the author). Shortly after the upgrade from Microsoft Office 2003 to Microsoft Office 2010, we noticed that the “Create a PDF” button was no longer functional in any Office application – nothing happened at all when we clicked it.
While our IT staff contacted both Microsoft and Adobe regarding this issue, my team began investigating workarounds to minimize the impact this would have on our publishing work. Unfortunately, we were only able to come up with two, less-than-stellar options.
Potential Solution #1 – Print to PDF
We quickly discovered that we could still create PDFs by printing to the Adobe PDF printer, but this option is far from ideal for documents destined for an eCTD submission. PDFs created using the Print function do not include any automatically created bookmarks or hyperlinks. Using this option would require us to manually create bookmarks and hyperlinks in every document. While this could be acceptable in limited circumstances, it’s certainly not a practical solution for producing eCTD submissions on a daily basis.
For More information about this article click here: eCTD Tips: Microsoft Office 2010 and PDF Creation Problems
The Problem
Prior to the upgrade, we used the “Create a PDF” button on the Acrobat toolbar in Word to create PDFs for submissions. As you know, PDFs created using this button will include functional bookmarks based upon the heading styles used, a fully hyperlinked table of contents, and functional internal hyperlinks within the text of the document (if they were created by the author). Shortly after the upgrade from Microsoft Office 2003 to Microsoft Office 2010, we noticed that the “Create a PDF” button was no longer functional in any Office application – nothing happened at all when we clicked it.
While our IT staff contacted both Microsoft and Adobe regarding this issue, my team began investigating workarounds to minimize the impact this would have on our publishing work. Unfortunately, we were only able to come up with two, less-than-stellar options.
Potential Solution #1 – Print to PDF
We quickly discovered that we could still create PDFs by printing to the Adobe PDF printer, but this option is far from ideal for documents destined for an eCTD submission. PDFs created using the Print function do not include any automatically created bookmarks or hyperlinks. Using this option would require us to manually create bookmarks and hyperlinks in every document. While this could be acceptable in limited circumstances, it’s certainly not a practical solution for producing eCTD submissions on a daily basis.
For More information about this article click here: eCTD Tips: Microsoft Office 2010 and PDF Creation Problems
15 June 2011
The Flow of Data from Bench to Bedside: Finally Enabled by Electronic Submission - By by Dirk Karsten Beth, President of Mission3, Inc
As with other industries, information is the substrate of Life Sciences, yet the Life Sciences have been slower to adopt information technology than other industries. Aircraft manufactures have been "virtually" test flying aircraft for decades now. The financial industry has been working with electronic regulatory submissions for about the same amount of time. But we are at a new age of life sciences, a period where discovery through bedside will be enhanced through the use of information technology.
The greater adoption of IT in Life Sciences is being driven not only through the clear benefits in quality and efficiency that we can all understand, but through market pressures and changes in the life sciences industry. Fewer blockbuster drugs and a higher failure rate in the discovery process; pharmacogenomics requires management of an ever increasing number of products in the portfolio; greater regulatory pressure; global development; biosimilar, biogeneric and generic pharmaceuticals are all affecting the way pharmaceutical and biotechnology companies operate. The response to this market pressure is a more efficient and effective industry — lead by information technology.
The true power of IT systems is first realized as the data flows freely from one system to the other and, as Life Sciences companies automate more processes internally, integration will lead to increased efficiency and quality.
For More Information Please us the weblink: The Flow of Data from Bench to Bedside: Finally Enabled by Electronic Submission.
The greater adoption of IT in Life Sciences is being driven not only through the clear benefits in quality and efficiency that we can all understand, but through market pressures and changes in the life sciences industry. Fewer blockbuster drugs and a higher failure rate in the discovery process; pharmacogenomics requires management of an ever increasing number of products in the portfolio; greater regulatory pressure; global development; biosimilar, biogeneric and generic pharmaceuticals are all affecting the way pharmaceutical and biotechnology companies operate. The response to this market pressure is a more efficient and effective industry — lead by information technology.
The true power of IT systems is first realized as the data flows freely from one system to the other and, as Life Sciences companies automate more processes internally, integration will lead to increased efficiency and quality.
For More Information Please us the weblink: The Flow of Data from Bench to Bedside: Finally Enabled by Electronic Submission.
Free Educational Webinar – New Rules in FDA Safety Reporting: Are You Prepared? - Download the Slides
Free Educational Webinar – New Rules in FDA Safety Reporting: Are You Prepared?
By Dr. Jack Snyder and Angela Overton of Cato Research 15 June 2011
Slides can be downloaded by clicking the weblink: Catco- New Rules in FDA Safety Reporting
By Dr. Jack Snyder and Angela Overton of Cato Research 15 June 2011
Slides can be downloaded by clicking the weblink: Catco- New Rules in FDA Safety Reporting
CDISC Live Free Webinar: CDISC Medical Device Standard - Your Change to Preview this New CDISC Standard
CDISC Live Webinar: CDISC Medical Device Standard - Your Change to Preview this New CDISC Standard
Friday 18 August 2011
11:00 AM Eastern / 10:00 AM Central
For more information and register for webinar please click: CDISC Live Free Webinar
Friday 18 August 2011
11:00 AM Eastern / 10:00 AM Central
For more information and register for webinar please click: CDISC Live Free Webinar
Submission Document Granularity - By Scott Cleve
Submission Document Granularity
by Scott Cleve, Director, Regulatory Affairs & Chris Buckley, Regulatory Affairs Associate Director at Astellas.
At its most basic level, increased document granularity within a submission simply means that there will be more documents to process and populate into a submission structure. More documents than what? - might be the next logical question. In the early years of electronic submissions most clinical study reports, were large and all inclusive of attachments and appendices. The drug product and drug substance documents were rolled up into complete files, respectively. Depending on the size of a study, a clinical study report document could exceed 100 MB in size and make the most confident publisher concerned about file corruption and poor performance. At that time, there were no requirements or expectations to sub-divide these documents into small defined granules by ICH or any regulatory authority.
Fast forward to the implementation of the Common Technical Document (CTD) and corresponding eCTD submission format that requires a greater degree of document granularity within a submission. With this emerging standard, the large documents needed to be authored as granular documents while still maintaining the overall content, research, and analysis of the entire topic (e.g. drug substance, drug product, clinical study). A benefit of the granulated approach is the ease of making changes or updates, review and approval, and lifecycle publishing of the granular document(s). On the publishing side, this concept eliminated the need to recreate bookmarks and hyperlinks in a large document and only perform these activities on the granular document(s). A challenge of maintaining granular documents is the need to conduct an impact analysis by the author and/or responsible department(s) to understand how a change to one granular document might impact other granular documents within the submission. A missing document in this analysis could result in an inconsistent message submitted to a regulatory authority, a problem that no sponsor company invites! Another challenge is maintaining the lifecycle links when certain destination documents are replaced or appended. Most current software cannot automatically update hyperlinks; therefore, potentially confusing the reviewer with outdated or broken links.
If you recall the basic concept of submission granularity of using a larger number of documents to create submissions, we have now complicated matters a bit. What was originally perceived as a submissions operations or publishing task is now an organizational, or at least a departmental process and/or strategy to develop granular content for submissions. It is not as simple as taking a large document and breaking it into smaller components. These components need to relate to each other and continue to provide continuity of the science and research of the submission application throughout its lifecycle. That is the entire organization’s responsibility, and not just the concern of the medical writing or publishing function. This change to a granular authoring approach may begin with the submissions operations or publishing group, but to successfully granulate documents the entire organization needs to be invested in developing granular content.
Source: GlobalSubmit Newsletter
http://globalsubmit.com
by Scott Cleve, Director, Regulatory Affairs & Chris Buckley, Regulatory Affairs Associate Director at Astellas.
At its most basic level, increased document granularity within a submission simply means that there will be more documents to process and populate into a submission structure. More documents than what? - might be the next logical question. In the early years of electronic submissions most clinical study reports, were large and all inclusive of attachments and appendices. The drug product and drug substance documents were rolled up into complete files, respectively. Depending on the size of a study, a clinical study report document could exceed 100 MB in size and make the most confident publisher concerned about file corruption and poor performance. At that time, there were no requirements or expectations to sub-divide these documents into small defined granules by ICH or any regulatory authority.
Fast forward to the implementation of the Common Technical Document (CTD) and corresponding eCTD submission format that requires a greater degree of document granularity within a submission. With this emerging standard, the large documents needed to be authored as granular documents while still maintaining the overall content, research, and analysis of the entire topic (e.g. drug substance, drug product, clinical study). A benefit of the granulated approach is the ease of making changes or updates, review and approval, and lifecycle publishing of the granular document(s). On the publishing side, this concept eliminated the need to recreate bookmarks and hyperlinks in a large document and only perform these activities on the granular document(s). A challenge of maintaining granular documents is the need to conduct an impact analysis by the author and/or responsible department(s) to understand how a change to one granular document might impact other granular documents within the submission. A missing document in this analysis could result in an inconsistent message submitted to a regulatory authority, a problem that no sponsor company invites! Another challenge is maintaining the lifecycle links when certain destination documents are replaced or appended. Most current software cannot automatically update hyperlinks; therefore, potentially confusing the reviewer with outdated or broken links.
If you recall the basic concept of submission granularity of using a larger number of documents to create submissions, we have now complicated matters a bit. What was originally perceived as a submissions operations or publishing task is now an organizational, or at least a departmental process and/or strategy to develop granular content for submissions. It is not as simple as taking a large document and breaking it into smaller components. These components need to relate to each other and continue to provide continuity of the science and research of the submission application throughout its lifecycle. That is the entire organization’s responsibility, and not just the concern of the medical writing or publishing function. This change to a granular authoring approach may begin with the submissions operations or publishing group, but to successfully granulate documents the entire organization needs to be invested in developing granular content.
Source: GlobalSubmit Newsletter
http://globalsubmit.com
14 June 2011
RAPS’ So. Cal. Workshops to Cover Increasingly Accepted Electronic Regulatory Submissions
RAPS’ So. Cal. Workshops to Cover Increasingly Accepted Electronic Regulatory Submissions
Regulatory agencies, including FDA and other global health regulators are increasingly accepting, and in some cases requiring, regulatory submissions in the electronic Common Technical Document (eCTD) format. The Regulatory Affairs Professionals Society (RAPS) will host two workshops on eCTD submissions in July in Long Beach, CA.
For More Information please click: RAPS Workshop eCTD
Regulatory agencies, including FDA and other global health regulators are increasingly accepting, and in some cases requiring, regulatory submissions in the electronic Common Technical Document (eCTD) format. The Regulatory Affairs Professionals Society (RAPS) will host two workshops on eCTD submissions in July in Long Beach, CA.
For More Information please click: RAPS Workshop eCTD
RPS: The Next Phase of eCTD (Regulated Product Submission Standard Increases Flexibility, Capabilities) By Joel Finkle
RPS: The Next Phase of eCTD
Regulated Product Submission standard increases flexibility, capabilities.
By Joel Finkle
June 8, 2011 | Guest Commentary | Today, almost two-thirds of all drug license application and renewal submissions are provided in the approved electronic Common Technical Document (eCTD) format. That figure rises to over 90 percent for electronic submissions to the U.S. Food and Drug Administration (FDA). This makes the job of the FDA much easier, allowing staff to call up and navigate their way through submissions quickly and logically. Applications are put together using meta tags and hyperlinks to attach related content in a way that makes the applications easily searchable.
Inevitably, eCTD has changed the way individuals and teams work, reducing bottlenecks and increasing accuracy by facilitating collaboration and parallel working, guided by clever software and even more clever business practices. In time, investments in compliant systems and processes will turn out to have been well worth it—especially when once-lengthy timescales for submission processing, reviewing, and approval are reduced.
In the same spirit, then, organizations should not be too alarmed to learn that there’s another new submission standard on the horizon.
For More Information Please use the link: BioIT World eCTD
Regulated Product Submission standard increases flexibility, capabilities.
By Joel Finkle
June 8, 2011 | Guest Commentary | Today, almost two-thirds of all drug license application and renewal submissions are provided in the approved electronic Common Technical Document (eCTD) format. That figure rises to over 90 percent for electronic submissions to the U.S. Food and Drug Administration (FDA). This makes the job of the FDA much easier, allowing staff to call up and navigate their way through submissions quickly and logically. Applications are put together using meta tags and hyperlinks to attach related content in a way that makes the applications easily searchable.
Inevitably, eCTD has changed the way individuals and teams work, reducing bottlenecks and increasing accuracy by facilitating collaboration and parallel working, guided by clever software and even more clever business practices. In time, investments in compliant systems and processes will turn out to have been well worth it—especially when once-lengthy timescales for submission processing, reviewing, and approval are reduced.
In the same spirit, then, organizations should not be too alarmed to learn that there’s another new submission standard on the horizon.
For More Information Please use the link: BioIT World eCTD
10 June 2011
EXTEDO Webinar - Managing the eRegulatory Submission
EXTEDO Webinar - Managing the eRegulatory Submission Lifecycle
Thursday, June 30, 2011 2:00 PM - 3:00 PM EDT
Please here to Register the Webinar: EXTEDO Webinar
Thursday, June 30, 2011 2:00 PM - 3:00 PM EDT
Please here to Register the Webinar: EXTEDO Webinar
EMA/TIGES issues Clarification Concerning the New Validation Criteria (3.1) for eCTD - Clarifies Pass Criteria

The Telematic Implementation Group for electronic submission and ICH Implementation(TIGes) has published additional clarification on the draft eCTD validation criteria 3.1 as published earlier this year (see our related News article).The additional clarification concerns the validation checks that require the presence of the previous eCTD lifecycle sequences to be performed correctly.
The first key message included in the clarification document is that "the revised criteria for technical validation of eCTD sequences (v3.1) should be treated as a single complete set of tests and not be divided in any way."
For More Information please click: EMA New Validation Criteria (3.1)
Swissmedic withdraws eCTD specification 1.01 and eCTD validation criteria 1.0
Swissmedic withdraws eCTD specification 1.01 and eCTD validation criteria 1.0
Please Click: Swissmedic eSubmissions Withdraws
Please Click: Swissmedic eSubmissions Withdraws
eCTD Office fully supports the creation of national e-submissions for Halmed/Croatia
eCTD Office - VNeeS Compiler - Full Support For VNeeS 2.0
(Veterinary) VNeeS Compiler, a part of eCTDOffice, is an off-the-shelf Windows software application for building error-free eSubmission for Veterinary Applications.
In March 2011, the TIGes Veterinary Subgroup has posted a new Guideline on eSubmissions for Veterinary Products. Guideline v.20 will come into effect on 01.09.2011.
Make sure September 2011 does not surprise you!
VNeeS Compiler fully supports the creation of VNeeS e-Submissions for pharmaceutical, immunological and MRL products. Both version 1.1 and recently published version 2.0 are supported.
VNeeS Compiler features:
Creates valid 1.1 and 2.0 Veterinary NeeS e-Submissions.
Creates VNeeS for a pharmaceutical, immunological or a MRL product.
Convert MS Word to PDF at export.
Splits / merges PDF documents.
Creates / auto-corrects PDF hyperlinks.
Generates GTOC and TOC files.
more reasons to choose VNeeS Compiler...
For More Information Please Click: eCTD Office
(Veterinary) VNeeS Compiler, a part of eCTDOffice, is an off-the-shelf Windows software application for building error-free eSubmission for Veterinary Applications.
In March 2011, the TIGes Veterinary Subgroup has posted a new Guideline on eSubmissions for Veterinary Products. Guideline v.20 will come into effect on 01.09.2011.
Make sure September 2011 does not surprise you!
VNeeS Compiler fully supports the creation of VNeeS e-Submissions for pharmaceutical, immunological and MRL products. Both version 1.1 and recently published version 2.0 are supported.
VNeeS Compiler features:
Creates valid 1.1 and 2.0 Veterinary NeeS e-Submissions.
Creates VNeeS for a pharmaceutical, immunological or a MRL product.
Convert MS Word to PDF at export.
Splits / merges PDF documents.
Creates / auto-corrects PDF hyperlinks.
Generates GTOC and TOC files.
more reasons to choose VNeeS Compiler...
For More Information Please Click: eCTD Office
TOPRA Regulatory Rapporteur: The implementation of eCTD in Japan
were as reference documents. However, the situation changed significantly in April 2009,when Japan’s Ministry of Health, Labour and Welfare (MHLW) issued a notification6 that paper dossiers were no longer required if applicants submitted the eCTD as the original dossier. Previously, the entire paper dossier was requested even if the eCTD was submitted as an original. Although paper Modules 1 and 2 are still required for review when the eCTD is submitted as the original dossier, it is no longer necessary to submit paper Modules 3, 4 and 5. As Figure 1 indicates, as the benefits of the eCTD have become better understood, the number of applications in eCTD format as the original dossier has increased.
For More Information please use the below link: TOPRA Regulatory Rapporteur: The implementation of eCTD in Japan
For More Information please use the below link: TOPRA Regulatory Rapporteur: The implementation of eCTD in Japan
07 June 2011
TheeCTDSummit: The Latest on Regulated Product Submission (RPS)
It has been a while since I have updated you on the RPS project. Back in September, I had expressed cautious optimism on being able to bring RPS to vote (ballot) in May 2011. We did not make the May 2011 ballot, thus the ballot is now delayed until September 2011.
We did not make the ballot, in my opinion, since we are still working on refining all of the requirements. We have two requirements left to be finalized; namely, being able to reuse studies and all of the components in two applications and support for the new European variation legislation.
Being able to reuse studies and all of the components in two applications will allow sponsors to create a study, define all of the components (e.g. protocol, study report body, synopsis, etc) in one application (e.g. investigational application) and refer to the study in your marketing application. This benefits sponsors since it allows them to synchronize content for a study in more than one application.
For More Information Please Click the Link:TheeCTDSummit: The Latest on Regulated Product Submission (RPS)
We did not make the ballot, in my opinion, since we are still working on refining all of the requirements. We have two requirements left to be finalized; namely, being able to reuse studies and all of the components in two applications and support for the new European variation legislation.
Being able to reuse studies and all of the components in two applications will allow sponsors to create a study, define all of the components (e.g. protocol, study report body, synopsis, etc) in one application (e.g. investigational application) and refer to the study in your marketing application. This benefits sponsors since it allows them to synchronize content for a study in more than one application.
For More Information Please Click the Link:TheeCTDSummit: The Latest on Regulated Product Submission (RPS)
04 June 2011
Microsoft Case Studies: Healthcare Company Expects Online Collaboration to Save Millions of Euros By Using eTMF
Healthcare Company Expects Online Collaboration to Save Millions of Euros
Sanofi is a multinational pharmaceutical company engaged in the research and development, manufacturing, and marketing of prescription drugs, vaccines, and over-the-counter healthcare products. Sanofi relied on time-consuming, paper-based processes to manage global drug trials. To introduce efficiencies and reduce costs, sanofi deployed a digitalized version of its previous method. Called the Clinical Trial Portal, the solution is based on the Clinical Portal solution from NextDocs that uses Microsoft portal, collaboration, and content management technologies. The new solution will accelerate time-to-market for new drug therapies and is expected to save an estimated 10,000 working days—costing €6.4 million (U.S.$9 million) in time wasted managing documents—when it is deployed to more than 3,000 internal and 40,000 external stakeholders over the next few years.
Situation
Sanofi has an extensive portfolio of prescription drugs, vaccines, generics, and consumer healthcare products targeting traditional markets and emerging countries. Sanofi addresses fundamental health issues and makes major therapeutic solutions available in its areas of expertise: oncology, diabetes, thrombotic and cardiovascular diseases, and central nervous system disorders. Sanofi Pasteur, the vaccines division of sanofi, offers vaccines protecting against 20 infectious diseases. Sales in 2010 for sanofi were €30,384 million (U.S. $43,967 million).
Goal to Streamline Drug Trials
To enable its strategic goals and build a more agile, adaptable organization, sanofi is improving efficiencies in its key business processes. The company began by focusing on how it brings its drugs to market. For sanofi, bringing a drug to market is a multiyear, complex process of global proportions. Expediting this process depends on the successful completion of clinical drug trials. At any given time, the company has dozens of trials in progress. For any trial, sanofi international clinical trial managers (CTMs) could be collaborating with affiliate hospitals or health facilities in as many as 50 countries, where clinical research associates (CRAs) at hundreds of local sites manage trial data from primary investigators, or physicians, administering the drug to many thousands of patients.
A Paper-Based Process
At sanofi, managing clinical drug trials was primarily a paper-based process. “Each clinical trial can generate more than 25,000 documents,” says Denis Cardinal, Domain Manager, Clinical Information and Collaboration, at sanofi. “These are collected during the course of the trial in a trial master file [TMF].”
At the beginning of a trial, when sanofi determines the feasibility of working with a local hospital and verifies that investigators have all the equipment and expertise to proceed, many documents travel between research sites and sanofi locations. However, there’s also a steady stream of documents sent back and forth in the intervening months or years of a trial. From start to finish, each document, and every subsequent revision to that document has to be collected locally, printed, signed, and sent to sanofi. There, sanofi quality control employees compile the TMF and store the documents in boxes to ensure that the company has an audit trail that shows how it complied with standard procedures every step of the way.
The logistics of managing this paper-based process for dozens of concurrent trials was daunting. Faxing, sending by courier, and emailing documents to sanofi was expensive and time consuming. Before participating in a clinical trial, physician investigators must sign a 1572 form required by the Food & Drug Administration (FDA). In addition, signed contracts and confidentiality agreements are required from CRAs at affiliate hospitals. Obtaining the original signatures on paper documents could take weeks. During the course of the trial, CTMs had to locate the right documents and ensure that they were up-to-date, which also incurred delays in the process.
Manual data gathering and file processing opened up the increased possibility of error; missing documents had to be found and entered into the TMF before the study could be officially closed. Also, with a paper-based document management system, it was very difficult to use and store metadata, such as authors, dates and other important contextual information. In the context of a long-term clinical trial, metadata is very important because it provides irrefutable evidence of who authorized, revised, and signed documents, and when. This information helps sanofi to follow industry regulations for clinical document management.
CTMs wanted an easy way to gain visibility into a trial’s progress instead of searching through the TMF or tracking down documents that might still be in transit. With trial documentation located in various places, it was difficult to ensure that clinical milestones were reached according to schedule and to communicate accurate progress reports to the team. Similarly, sanofi executives did not have an up-to-the minute, global view of all trials underway.
“We needed to replace an outdated paper-based process and align ourselves with the health industry’s push towards electronic data exchange that adheres to the latest regulations on security and privacy,”
says Cardinal. “We envisioned a web-based portal solution that would encompass sophisticated document management capabilities, including digital signatures, and a global framework to enable internal and external stakeholders to collaborate and communicate efficiently on clinical trials. The solution had to provide easy access to clinical trial documents and applications and a global view of each study’s progress.”
Solution
After evaluating different document management and collaboration solutions on the market, sanofi narrowed its decision down to two vendors: Microsoft and a leading competitor. As part of its evaluation, sanofi invited Microsoft Managed Partner NextDocs to visit the Paris headquarters and conduct a proof-of-concept (POC). NextDocs has expertise in building SharePoint-based compliance solutions that address regulatory requirements in the healthcare industry. Sanofi was interested to see how the NextDocs Clinical Portal solution worked with Microsoft Office SharePoint Server 2007 to fulfill the company’s requirements for an easy-to-use health portal and compliant document management solution.
The Clinical Trial Portal built on Microsoft technologies enables efficient global collaboration that directly supports our ambition to … sit among the top clinical development performers.
Fabien Jolly
Vice President,
Technology and Information Management,
sanofi.
“When we saw SharePoint Server in action with the NextDocs Clinical Portal solution, which ensures industry regulatory compliance, we were won over by the ease of use, adaptability, and additional functionality of the technologies,”
says Cardinal. “SharePoint Server offers much more than document management capabilities: it’s a scalable, user-friendly portal and collaboration platform with additional features such as subscriptions, notifications, shared calendars, and message boards. We had already deployed Office SharePoint Server 2007 for a few internal collaboration sites and we appreciated the value of taking advantage of existing IT capabilities.”
Sanofi also chose Office SharePoint Server 2007 because it could easily interoperate with existing line-of-business solutions, including the Clinical Trial Management System (CTMS) and an Oracle-based solution that tracks employee permissions to access corporate network resources. Also, the company liked the product’s familiar, easy-to-use interface. The interoperability between Office SharePoint Server 2007 and Microsoft Office desktop productivity software would ensure a ready adoption of the solution for clinical research associates and investigators because they could access and store documents within the familiar Office business applications that they rely on to create and modify content.
Requirements Gathered
Beginning in June 2009, sanofi began gathering high-level requirements from CTMs, hospital staff, doctors, and CRAs around the world. This tremendous undertaking was necessary to ensure the creation of a digital system that improved upon the manual process. At the height of the project, there were 40 people from sanofi involved in different project areas such as trial contact management, integration with external systems, and building standardized taxonomy and metadata.
“With our new clinical trial portal, we were determined to design and implement optimized processes and to standardize document metadata,” says Cardinal. “We defined 650 different document types. All documents are tagged with clinical keywords defined by the team so we can use SharePoint search functionality instead of looking through paper files. All the document templates were set up using the same phrases and keywords throughout the company so that documentation will be standard and familiar to everyone.”
Instead of sending paper documents to be filed in the TMF in boxes at sanofi locations, internal and external stakeholders will upload documents to the Clinical Trial Portal to create an electronic Trial Master File, (eTMF) that stores documents and all associated updates online. The difference is that now, based on user access and permissions, internal and external international team members will be able to see the contents of the eTMF and watch carefully as documents are added and milestones are reached.
Security Concerns Addressed
As sanofi makes the transition to a digital clinical trial management process and electronic text documents replace paper records, it must comply with government and industry regulations that govern the flow of digital information. Working with NextDocs, sanofi added electronic and digital signature capabilities and new workflows that comply with Title 21 CFR Part 11 containing FDA guidelines on electronic records. Now an investigator can file a 1572 form online using a digital signature, which is a coded message unique to both the document and the signer. Sanofi has 30 days to process these forms, so in order to stay on schedule a SharePoint-based workflow sends a reminder email message to the correct recipient’s Microsoft Office Outlook inbox or mobile phone using Microsoft Exchange ActiveSync technology.
NextDocs used SharePoint Timer jobs to ensure that the Clinical Trial Portal module interoperates with the CTMS to synchronize portal data with the information contained in the CTMS and an Oracle-based permissions system. This makes it easy for sanofi staff to share appropriate study and management data from the CTMS system with trial participants around the world. For example, trials with an affiliate hospital must include a signed confidentiality agreement that resides in the CTMS. For the link to the permissions system, NextDocs developers simply replicated existing permissions when assigning users to the Clinical Trial Portal, which ensures that everyone continues to access the information for which they have permission, according to responsibility and position.
Designed to Scale
Sanofi is in the final stages of deploying the Clinical Trial Portal based on Office SharePoint Server 2007, running the solution in the test and development environment to correct any last-minute issues. The company released the first live version of the Clinical Trial Portal in April 2011 and by May 2011 two new studies were using the system. Another two new studies are also scheduled to start using the system in May 2011. NextDocs offers a version of its clinical health portal that takes advantage of Microsoft SharePoint Server 2010 and sanofi will be upgrading its Clinical Trial Portal to SharePoint Server 2010 at the beginning of 2012.
While the first live studies will focus on two small trials, sanofi designed the Clinical Trial Portal to scale to thousands of users and has high expectations for performance and reliability. “When we are fully deployed, we’re expecting to reach approximately 3,000 internal users, including affiliate staff at up to 10,000 hospital sites, where between 30,000 and 40,000 doctors will be gathering and uploading clinical trial data into the portal,” says Cardinal. “We worked with Microsoft to complete two performance tests to ensure our SharePoint environment is well-configured and scalable. We’re confident that the clinical portal solution will perform well. Using the system requires little training, encouraging a quick adoption rate and positive user response.”
“With the new Clinical Trial Portal, we leverage best-in-class portal and document management technologies to produce a rich user experience and enable efficient global collaboration,” adds Fabien Jolly, Vice President of Technology and Information Management at sanofi.
Benefits
Thanks to Microsoft collaboration technologies, sanofi is poised to experience a paradigm shift in clinical trial management and operational efficiency. Replacing a manual, paper-intensive process with a web-based digital solution was a daunting project. However, the company benefitted from the Microsoft partner ecosystem by taking advantage of NextDocs’ expertise in extending SharePoint technologies to meet its needs. The end result combined health portal capabilities with compliant document management to deliver an innovative solution that will speed time-to-market, improve productivity, reduce costs and risk, and drive a competitive advantage.
“In a networked, open source environment, the Clinical Trial Portal leverages the capabilities for integrated technologies, improves the productivity, speed, and quality of our trials, and liberates the investigators to focus on the care of the patients,” says Ji Zhang, Vice President, Head of Clinical Sciences and Operations Worldwide, sanofi.
Increased Insight Improves Trial Management
Instead of waiting until the end of a trial to ensure that all required, signed documentation is included in the trial master file, CTMs will be able to monitor which documents are added and when, taking steps along the way to submit the proper documentation to applicable regulatory agencies on time and to ensure that nothing is missing before going to the next step.
With the new Clinical Trial Portal, we leverage best-in-class portal and document management technologies to produce a rich user experience and enable efficient global collaboration.
Fabien Jolly
Vice President,
Technology and Information Management,
sanofi
“With real-time document submission to the Clinical Trial Portal, trial managers can measure the performance of their teams to see if they are providing the right information,” says Cardinal. “And with aggregate trial data available, sanofi executives will gain a new level of insight into global operations.”
Seamless Collaboration Drives Productivity
The Clinical Trial Portal provides easy access to one platform so that all internal and external stakeholders will be able to collaborate and communicate efficiently on drug trials no matter where they are in the world. The solution not only delivers streamlined information sharing for clinical trial data, but also facilitates access to data from back-end clinical management and user permission applications.
If one team member adds a document, or updates a file, everyone else on the team will be instantly aware of the change and can work with the most recent draft. With everything located in one easily accessed location, sanofi estimates that document versions will be reduced by 30 percent.
“Instead of phoning and emailing clinical trial team members, our stakeholders will be able to work together more efficiently, responding to issues and delivering results more quickly,” says Cardinal. “When an investigator connects to the portal, he or she will see all the documents in the electronic Trial Master File and all contact information for his or her colleagues throughout the trial. Easy communication and collaboration builds teamwork and improves productivity all around.”
Digital Collaboration Reduces Costs
After sanofi rolls out the Clinical Trial Portal globally, the company will be saving a significant amount of money on long-distance phone bills and fax, courier, and paper costs. “It’s easy to imagine the savings in labor costs if you think about the time and effort required to manage 25,000 documents, and that’s just for one trial,” says Cardinal. “Even if we take into account the cost of maintaining the new system, we still expect to experience a positive return on investment within the first 2 years.”
As an example, for a single study the average trial master file contains up to 25,000 documents. Conducting 100 trials per year requires the creation, review/approval, and management of up to 2,500,000 documents per year. “If we consider that for each document lifecycle we could save only 2 minutes—a very conservative example—in searching, processing, and posting documents, it adds up to 5,000,000 minutes, or approximately 10,000 person-days of time savings per year,” says Cardinal. “At a conservative average hourly wage of €80 for the sanofi employees involved in managing documents for the clinical trials, this could add up to a savings of €6.4 million in labor costs when the solution is fully deployed over the next few years. Now the trial stakeholders can focus on additional added-value or core medical activities.”
Faster Time-to-Market Improves Public Health
The Clinical Portal Solution provides a communication forum for users to share their views about a clinical trial in real time. It used to take several weeks to obtain signed contracts before launching a trial, but now this step will be accomplished in minutes, thanks to the digital signature function in the Clinical Trial Portal. And CTMs and quality assurance staff will no longer be spending days sorting through the TMF to make sure it contains all of the documents required by regulatory agencies: this task will be accomplished at regular intervals as documents are uploaded during the course of a trial, saving a significant amount of time.
“The Clinical Trial Portal will simplify the day-to-day activities of thousands of people that work in our clinical trials,” says Marie Sebille, Vice President of Trial Operations at sanofi. “This will help us gain time during the startup and completion of a trial, which is one of our major challenges.”
“We expect the Clinical Trial Portal to help us bring drugs to market faster,” adds Cardinal. “That’s because everyone will be working in a secure, compliant environment where it’s easy to create, store, and find the documents they need, when they need them.“
Regulatory Compliance Features Reduced Risk
The Clinical Portal Solution will make it easier for sanofi to satisfy regulatory compliance requirements by providing audit trails, electronic signatures, strict access control, accurate copies, and proper storage of all document versions.
“In the pharmaceutical industry, we can’t negotiate with quality control,” says Cardinal. “Now that we have metadata specifying the author and revision history for each document, we will be able to track and audit every transaction.”
Innovation Builds Competitive Advantage
With its Clinical Trial Portal based on Microsoft technologies, sanofi has moved to the forefront of drug trial management innovation. When it rolls out the solution worldwide, sanofi expects positive feedback from clinical research associates and investigators working in the field. “We appreciate that for our investigators this is additional work to caring for their patients, so we want to make it as easy as possible to work for sanofi,” says Cardinal. “The Clinical Trial Portal will be an important tool that we can use to differentiate our company and keep our investigators happy, because they are essential to the process.”
Eventually, sanofi could use the Clinical Trial Portal to forge partnerships with other organizations, a key element of the company’s evolving business model. “This portal is truly welcomed by the clinical study units,” adds Mark Travers, Vice President, Clinical Study Units at sanofi. “We see it as a strategic vehicle where sanofi and our partners can exchange information in real time and as the interface of our organization with the clinical research community.”
“The Clinical Trial Portal built on Microsoft technologies enables efficient global collaboration that directly supports our ambition to be a sponsor of choice for clinical trials and to sit among the top clinical development performers,” concludes Jolly.
Microsoft Solutions for the Healthcare Industry
Healthcare and life sciences organizations are under tremendous pressure to meet regulatory requirements, improve patient care, and reduce the time it takes to develop drugs and take them to market. To meet this challenge, Microsoft and its partners have developed cost-effective solutions that enable healthcare organizations to streamline and automate daily processes that improve productivity and deliver information whenever and wherever it is needed. The result is enhanced productivity, safety, and quality.
For more information about Microsoft solutions for the healthcare industry, go to:
www.microsoft.com/healthcare
Sanofi is a multinational pharmaceutical company engaged in the research and development, manufacturing, and marketing of prescription drugs, vaccines, and over-the-counter healthcare products. Sanofi relied on time-consuming, paper-based processes to manage global drug trials. To introduce efficiencies and reduce costs, sanofi deployed a digitalized version of its previous method. Called the Clinical Trial Portal, the solution is based on the Clinical Portal solution from NextDocs that uses Microsoft portal, collaboration, and content management technologies. The new solution will accelerate time-to-market for new drug therapies and is expected to save an estimated 10,000 working days—costing €6.4 million (U.S.$9 million) in time wasted managing documents—when it is deployed to more than 3,000 internal and 40,000 external stakeholders over the next few years.
Situation
Sanofi has an extensive portfolio of prescription drugs, vaccines, generics, and consumer healthcare products targeting traditional markets and emerging countries. Sanofi addresses fundamental health issues and makes major therapeutic solutions available in its areas of expertise: oncology, diabetes, thrombotic and cardiovascular diseases, and central nervous system disorders. Sanofi Pasteur, the vaccines division of sanofi, offers vaccines protecting against 20 infectious diseases. Sales in 2010 for sanofi were €30,384 million (U.S. $43,967 million).
Goal to Streamline Drug Trials
To enable its strategic goals and build a more agile, adaptable organization, sanofi is improving efficiencies in its key business processes. The company began by focusing on how it brings its drugs to market. For sanofi, bringing a drug to market is a multiyear, complex process of global proportions. Expediting this process depends on the successful completion of clinical drug trials. At any given time, the company has dozens of trials in progress. For any trial, sanofi international clinical trial managers (CTMs) could be collaborating with affiliate hospitals or health facilities in as many as 50 countries, where clinical research associates (CRAs) at hundreds of local sites manage trial data from primary investigators, or physicians, administering the drug to many thousands of patients.
A Paper-Based Process
At sanofi, managing clinical drug trials was primarily a paper-based process. “Each clinical trial can generate more than 25,000 documents,” says Denis Cardinal, Domain Manager, Clinical Information and Collaboration, at sanofi. “These are collected during the course of the trial in a trial master file [TMF].”
At the beginning of a trial, when sanofi determines the feasibility of working with a local hospital and verifies that investigators have all the equipment and expertise to proceed, many documents travel between research sites and sanofi locations. However, there’s also a steady stream of documents sent back and forth in the intervening months or years of a trial. From start to finish, each document, and every subsequent revision to that document has to be collected locally, printed, signed, and sent to sanofi. There, sanofi quality control employees compile the TMF and store the documents in boxes to ensure that the company has an audit trail that shows how it complied with standard procedures every step of the way.
The logistics of managing this paper-based process for dozens of concurrent trials was daunting. Faxing, sending by courier, and emailing documents to sanofi was expensive and time consuming. Before participating in a clinical trial, physician investigators must sign a 1572 form required by the Food & Drug Administration (FDA). In addition, signed contracts and confidentiality agreements are required from CRAs at affiliate hospitals. Obtaining the original signatures on paper documents could take weeks. During the course of the trial, CTMs had to locate the right documents and ensure that they were up-to-date, which also incurred delays in the process.
Manual data gathering and file processing opened up the increased possibility of error; missing documents had to be found and entered into the TMF before the study could be officially closed. Also, with a paper-based document management system, it was very difficult to use and store metadata, such as authors, dates and other important contextual information. In the context of a long-term clinical trial, metadata is very important because it provides irrefutable evidence of who authorized, revised, and signed documents, and when. This information helps sanofi to follow industry regulations for clinical document management.
CTMs wanted an easy way to gain visibility into a trial’s progress instead of searching through the TMF or tracking down documents that might still be in transit. With trial documentation located in various places, it was difficult to ensure that clinical milestones were reached according to schedule and to communicate accurate progress reports to the team. Similarly, sanofi executives did not have an up-to-the minute, global view of all trials underway.
“We needed to replace an outdated paper-based process and align ourselves with the health industry’s push towards electronic data exchange that adheres to the latest regulations on security and privacy,”
says Cardinal. “We envisioned a web-based portal solution that would encompass sophisticated document management capabilities, including digital signatures, and a global framework to enable internal and external stakeholders to collaborate and communicate efficiently on clinical trials. The solution had to provide easy access to clinical trial documents and applications and a global view of each study’s progress.”
Solution
After evaluating different document management and collaboration solutions on the market, sanofi narrowed its decision down to two vendors: Microsoft and a leading competitor. As part of its evaluation, sanofi invited Microsoft Managed Partner NextDocs to visit the Paris headquarters and conduct a proof-of-concept (POC). NextDocs has expertise in building SharePoint-based compliance solutions that address regulatory requirements in the healthcare industry. Sanofi was interested to see how the NextDocs Clinical Portal solution worked with Microsoft Office SharePoint Server 2007 to fulfill the company’s requirements for an easy-to-use health portal and compliant document management solution.
The Clinical Trial Portal built on Microsoft technologies enables efficient global collaboration that directly supports our ambition to … sit among the top clinical development performers.
Fabien Jolly
Vice President,
Technology and Information Management,
sanofi.
“When we saw SharePoint Server in action with the NextDocs Clinical Portal solution, which ensures industry regulatory compliance, we were won over by the ease of use, adaptability, and additional functionality of the technologies,”
says Cardinal. “SharePoint Server offers much more than document management capabilities: it’s a scalable, user-friendly portal and collaboration platform with additional features such as subscriptions, notifications, shared calendars, and message boards. We had already deployed Office SharePoint Server 2007 for a few internal collaboration sites and we appreciated the value of taking advantage of existing IT capabilities.”
Sanofi also chose Office SharePoint Server 2007 because it could easily interoperate with existing line-of-business solutions, including the Clinical Trial Management System (CTMS) and an Oracle-based solution that tracks employee permissions to access corporate network resources. Also, the company liked the product’s familiar, easy-to-use interface. The interoperability between Office SharePoint Server 2007 and Microsoft Office desktop productivity software would ensure a ready adoption of the solution for clinical research associates and investigators because they could access and store documents within the familiar Office business applications that they rely on to create and modify content.
Requirements Gathered
Beginning in June 2009, sanofi began gathering high-level requirements from CTMs, hospital staff, doctors, and CRAs around the world. This tremendous undertaking was necessary to ensure the creation of a digital system that improved upon the manual process. At the height of the project, there were 40 people from sanofi involved in different project areas such as trial contact management, integration with external systems, and building standardized taxonomy and metadata.
“With our new clinical trial portal, we were determined to design and implement optimized processes and to standardize document metadata,” says Cardinal. “We defined 650 different document types. All documents are tagged with clinical keywords defined by the team so we can use SharePoint search functionality instead of looking through paper files. All the document templates were set up using the same phrases and keywords throughout the company so that documentation will be standard and familiar to everyone.”
Instead of sending paper documents to be filed in the TMF in boxes at sanofi locations, internal and external stakeholders will upload documents to the Clinical Trial Portal to create an electronic Trial Master File, (eTMF) that stores documents and all associated updates online. The difference is that now, based on user access and permissions, internal and external international team members will be able to see the contents of the eTMF and watch carefully as documents are added and milestones are reached.
Security Concerns Addressed
As sanofi makes the transition to a digital clinical trial management process and electronic text documents replace paper records, it must comply with government and industry regulations that govern the flow of digital information. Working with NextDocs, sanofi added electronic and digital signature capabilities and new workflows that comply with Title 21 CFR Part 11 containing FDA guidelines on electronic records. Now an investigator can file a 1572 form online using a digital signature, which is a coded message unique to both the document and the signer. Sanofi has 30 days to process these forms, so in order to stay on schedule a SharePoint-based workflow sends a reminder email message to the correct recipient’s Microsoft Office Outlook inbox or mobile phone using Microsoft Exchange ActiveSync technology.
NextDocs used SharePoint Timer jobs to ensure that the Clinical Trial Portal module interoperates with the CTMS to synchronize portal data with the information contained in the CTMS and an Oracle-based permissions system. This makes it easy for sanofi staff to share appropriate study and management data from the CTMS system with trial participants around the world. For example, trials with an affiliate hospital must include a signed confidentiality agreement that resides in the CTMS. For the link to the permissions system, NextDocs developers simply replicated existing permissions when assigning users to the Clinical Trial Portal, which ensures that everyone continues to access the information for which they have permission, according to responsibility and position.
Designed to Scale
Sanofi is in the final stages of deploying the Clinical Trial Portal based on Office SharePoint Server 2007, running the solution in the test and development environment to correct any last-minute issues. The company released the first live version of the Clinical Trial Portal in April 2011 and by May 2011 two new studies were using the system. Another two new studies are also scheduled to start using the system in May 2011. NextDocs offers a version of its clinical health portal that takes advantage of Microsoft SharePoint Server 2010 and sanofi will be upgrading its Clinical Trial Portal to SharePoint Server 2010 at the beginning of 2012.
While the first live studies will focus on two small trials, sanofi designed the Clinical Trial Portal to scale to thousands of users and has high expectations for performance and reliability. “When we are fully deployed, we’re expecting to reach approximately 3,000 internal users, including affiliate staff at up to 10,000 hospital sites, where between 30,000 and 40,000 doctors will be gathering and uploading clinical trial data into the portal,” says Cardinal. “We worked with Microsoft to complete two performance tests to ensure our SharePoint environment is well-configured and scalable. We’re confident that the clinical portal solution will perform well. Using the system requires little training, encouraging a quick adoption rate and positive user response.”
“With the new Clinical Trial Portal, we leverage best-in-class portal and document management technologies to produce a rich user experience and enable efficient global collaboration,” adds Fabien Jolly, Vice President of Technology and Information Management at sanofi.
Benefits
Thanks to Microsoft collaboration technologies, sanofi is poised to experience a paradigm shift in clinical trial management and operational efficiency. Replacing a manual, paper-intensive process with a web-based digital solution was a daunting project. However, the company benefitted from the Microsoft partner ecosystem by taking advantage of NextDocs’ expertise in extending SharePoint technologies to meet its needs. The end result combined health portal capabilities with compliant document management to deliver an innovative solution that will speed time-to-market, improve productivity, reduce costs and risk, and drive a competitive advantage.
“In a networked, open source environment, the Clinical Trial Portal leverages the capabilities for integrated technologies, improves the productivity, speed, and quality of our trials, and liberates the investigators to focus on the care of the patients,” says Ji Zhang, Vice President, Head of Clinical Sciences and Operations Worldwide, sanofi.
Increased Insight Improves Trial Management
Instead of waiting until the end of a trial to ensure that all required, signed documentation is included in the trial master file, CTMs will be able to monitor which documents are added and when, taking steps along the way to submit the proper documentation to applicable regulatory agencies on time and to ensure that nothing is missing before going to the next step.
With the new Clinical Trial Portal, we leverage best-in-class portal and document management technologies to produce a rich user experience and enable efficient global collaboration.
Fabien Jolly
Vice President,
Technology and Information Management,
sanofi
“With real-time document submission to the Clinical Trial Portal, trial managers can measure the performance of their teams to see if they are providing the right information,” says Cardinal. “And with aggregate trial data available, sanofi executives will gain a new level of insight into global operations.”
Seamless Collaboration Drives Productivity
The Clinical Trial Portal provides easy access to one platform so that all internal and external stakeholders will be able to collaborate and communicate efficiently on drug trials no matter where they are in the world. The solution not only delivers streamlined information sharing for clinical trial data, but also facilitates access to data from back-end clinical management and user permission applications.
If one team member adds a document, or updates a file, everyone else on the team will be instantly aware of the change and can work with the most recent draft. With everything located in one easily accessed location, sanofi estimates that document versions will be reduced by 30 percent.
“Instead of phoning and emailing clinical trial team members, our stakeholders will be able to work together more efficiently, responding to issues and delivering results more quickly,” says Cardinal. “When an investigator connects to the portal, he or she will see all the documents in the electronic Trial Master File and all contact information for his or her colleagues throughout the trial. Easy communication and collaboration builds teamwork and improves productivity all around.”
Digital Collaboration Reduces Costs
After sanofi rolls out the Clinical Trial Portal globally, the company will be saving a significant amount of money on long-distance phone bills and fax, courier, and paper costs. “It’s easy to imagine the savings in labor costs if you think about the time and effort required to manage 25,000 documents, and that’s just for one trial,” says Cardinal. “Even if we take into account the cost of maintaining the new system, we still expect to experience a positive return on investment within the first 2 years.”
As an example, for a single study the average trial master file contains up to 25,000 documents. Conducting 100 trials per year requires the creation, review/approval, and management of up to 2,500,000 documents per year. “If we consider that for each document lifecycle we could save only 2 minutes—a very conservative example—in searching, processing, and posting documents, it adds up to 5,000,000 minutes, or approximately 10,000 person-days of time savings per year,” says Cardinal. “At a conservative average hourly wage of €80 for the sanofi employees involved in managing documents for the clinical trials, this could add up to a savings of €6.4 million in labor costs when the solution is fully deployed over the next few years. Now the trial stakeholders can focus on additional added-value or core medical activities.”
Faster Time-to-Market Improves Public Health
The Clinical Portal Solution provides a communication forum for users to share their views about a clinical trial in real time. It used to take several weeks to obtain signed contracts before launching a trial, but now this step will be accomplished in minutes, thanks to the digital signature function in the Clinical Trial Portal. And CTMs and quality assurance staff will no longer be spending days sorting through the TMF to make sure it contains all of the documents required by regulatory agencies: this task will be accomplished at regular intervals as documents are uploaded during the course of a trial, saving a significant amount of time.
“The Clinical Trial Portal will simplify the day-to-day activities of thousands of people that work in our clinical trials,” says Marie Sebille, Vice President of Trial Operations at sanofi. “This will help us gain time during the startup and completion of a trial, which is one of our major challenges.”
“We expect the Clinical Trial Portal to help us bring drugs to market faster,” adds Cardinal. “That’s because everyone will be working in a secure, compliant environment where it’s easy to create, store, and find the documents they need, when they need them.“
Regulatory Compliance Features Reduced Risk
The Clinical Portal Solution will make it easier for sanofi to satisfy regulatory compliance requirements by providing audit trails, electronic signatures, strict access control, accurate copies, and proper storage of all document versions.
“In the pharmaceutical industry, we can’t negotiate with quality control,” says Cardinal. “Now that we have metadata specifying the author and revision history for each document, we will be able to track and audit every transaction.”
Innovation Builds Competitive Advantage
With its Clinical Trial Portal based on Microsoft technologies, sanofi has moved to the forefront of drug trial management innovation. When it rolls out the solution worldwide, sanofi expects positive feedback from clinical research associates and investigators working in the field. “We appreciate that for our investigators this is additional work to caring for their patients, so we want to make it as easy as possible to work for sanofi,” says Cardinal. “The Clinical Trial Portal will be an important tool that we can use to differentiate our company and keep our investigators happy, because they are essential to the process.”
Eventually, sanofi could use the Clinical Trial Portal to forge partnerships with other organizations, a key element of the company’s evolving business model. “This portal is truly welcomed by the clinical study units,” adds Mark Travers, Vice President, Clinical Study Units at sanofi. “We see it as a strategic vehicle where sanofi and our partners can exchange information in real time and as the interface of our organization with the clinical research community.”
“The Clinical Trial Portal built on Microsoft technologies enables efficient global collaboration that directly supports our ambition to be a sponsor of choice for clinical trials and to sit among the top clinical development performers,” concludes Jolly.
Microsoft Solutions for the Healthcare Industry
Healthcare and life sciences organizations are under tremendous pressure to meet regulatory requirements, improve patient care, and reduce the time it takes to develop drugs and take them to market. To meet this challenge, Microsoft and its partners have developed cost-effective solutions that enable healthcare organizations to streamline and automate daily processes that improve productivity and deliver information whenever and wherever it is needed. The result is enhanced productivity, safety, and quality.
For more information about Microsoft solutions for the healthcare industry, go to:
www.microsoft.com/healthcare
IIR :eCTD Seminar in Spain:"ECTD Electronic Drug Registration up to date" - Barcelona, October 4, 2011
IIR :eCTD Seminar in Spain:"ECTD Electronic Drug Registration up to date" - Barcelona, October 4, 2011
For More Information Please click the link: eCTD Seminar in Spain
For More Information Please click the link: eCTD Seminar in Spain
02 June 2011
Article: The Challenges of e-Publishing in Regulatory Affairs in online journal International Pharmaceutical Industry
Article: The Challenges of e-Publishing in Regulatory Affairs in online journal International Pharmaceutical Industry
Please click the link to find the Article: The Challenges of e-Publishing in Regulatory Affairs
Please click the link to find the Article: The Challenges of e-Publishing in Regulatory Affairs
ECTD: A Business Case for Small to Midsize Companies - presentation from Monte Levinson
ECTD: A Business Case for Small to Midsize Companies - presentation from Monte Levinson
Please click the link to find the presentation: ECTD: A Business Case for Small to Midsize Companies
Please click the link to find the presentation: ECTD: A Business Case for Small to Midsize Companies
Health Canada: Drug Submission Performance Reports

he Drug Submission Review Performance Reports provide detailed metrics about the timeliness of pre-market drug review process against the performance service standards. The annual report compares five calendar years, while the quarterly report compares five quarters. The reports are broken down by operational areas. The Therapeutic Product (TPD) Report covers pharmaceuticals, while the Biologic and Therapeutic Products Directorate (BGTD) Report covers biologics. Within each report, statistics are provided by submission type and show the number received, the number in workload, the number of decisions and the number of approvals and time to approval.
Submissions Received are counts of submissions received during the year using from the filing date. Workload is reported as the number of submissions "under active review" on a given day. "Backlog" is the proportion of the workload that is over target. Approvals are Notice of Compliances (NOC) issued or issuable. A NOC issuable is when a submission's NOC is placed "on hold" awaiting authorization to market, due to Patent regulations or due to de-scheduling (from prescription to Over the Counter).
For More Information please Click: Drug Submission Performance Reports
01 June 2011
HMA & EMA: More Transparancy in Application Dossiers - DRAFT FOR PUBLIC CONSULTATION

Document on the Identification of Commercially Confidential Information and Protection of personal Data within the structure of the marketing Authorisation (MA) dossier release of Information after granting of a marketing Authorisation
The Heads of Medicines Agencies (HMA) and European Medicines Agency (EMA) havebeen working together with a view towards achieving greater transparency of their operations and better addressing the increasing requests for information theyfrom members of the civil society.
This draft guidance document is presented as a consensus document agreed by theentire Network of Competent Authorities, laying down practical orientations for national and European authorities in regards to requests for access to information contained in MA dossiers.
For More Information Please Click: HMA & EMA: More Transparancy in Application Dossiers - DRAFT FOR PUBLIC CONSULTATION
European Medicines Agency and Heads of Medicines Agencies propose measures to make information in application dossiers more transparent

The European Medicines Agency and the Heads of Medicines Agencies (HMA) have released a guidance document on the identification of commercially confidential information and protection of personal data within the structure of the marketing-authorisation dossier for public consultation.
The draft document, which is open for comment until 1 September 2011, outlines the types of information included in marketing-authorisation applications that can be released following a request for access to documents, once a marketing authorisation has been granted.
The consultation is being conducted to gather the views of stakeholders, including pharmaceutical companies, healthcare professionals and patient organisations. The Agency and the HMA are particularly interested in receiving comments on the criteria for the release or protection of personal data, whether contractual arrangements between companies need to be maintained as confidential, and how to address the personal security of individuals involved in studies using animals. They are also inviting stakeholders to make proposals on how to make these types of request easier.
For more Information please click: EMA Guidance Document for protection of personal data
DIA Webinar: Electronic Submission Basics Webinar Series: Part 2- eCTD 101: Concepts of the eCTD Standard Sep 22 2011
WEBINAR: Electronic Submission Basics: 3-part Webinar Series: Part 2- eCTD 101: Concepts of the eCTD Standard
Date(s) And Time(s):
Sep 22 2011 11:00AM - Sep 22 2011 12:30PM
Interest Area(s):
Regulatory Affairs,Project Management,Medical Writing,IT/Validation,Clinical Research,Clinical Data Management/ eClinical
Overview:
Register ONLINE in ONE transaction for
MULTIPLE Webinars in this SERIES and SAVE UP TO 15%!*
*Special Pricing Valid only on Online Registrations for Both Individuals and Group Sites!
You may also register for a single webinar (at no discount) online or by fax. See Page 4 of the Series Program for the registration form.
For More Information Please Click: DIA eSubmission Basics Webinar Series
EMA Draft Concept To Revise CTD Guideline For Traditional Herbal Medicinal Products

Concept paper on the revision of the guideline on the use of the CTD format in the preparation of a registration application for traditional herbal medicinal products - Focus on M3 quality
For More information Please Click: EMA - Revise CTD Guideline For Traditional Herbal Medicinal Products
Exalon:New status reports for the European eSubmission Gateway and electronic Application Form project released

The European Medicines Agency has released new status reports regarding the European eSubmission Gateway and the electronic Application Form (eAF) projects. Both status reports have the report date "June 2011" and are available for download from the "eSubmission" website of the Agency.
Short summary eSubmission Gateway Project:
The eSubmission Gateway will use Axway technology to allow registered business partners to send eCTD submissions electronically to the EMA via the gateway. Progress of the project has been delayed for several months due to the resignation of the project manager and subsequently the entire development and test team (see previous status report from March 2011).
According to the new status report the business requirements and software architecture documentation for release 1 of the gateway are final and development test phase have been completed. System testing is currently ongoing and will be completed in June followed by the user acceptance testing.
Modified work instructions for the handling of incoming eCTD submissions and details about the registration process will be published in July 2011.
The pilot phase is planned to start also in July 2011 whereas the start of the production phase of Release 1 of the eSubmission Gateway is planned for October 2011. The Agency expects that "that both methods to submit eCTDs (via CDs/DVDs and via eSubmission Gateway) will be supported for some time"
For More Information Please Click: Exalon: New Status Reports for ESG
Ask Cato: Health Canada Expands Acceptance of eCTD Submissions
Last week, Health Canada (HC) issued a Notice to to announce an increase in the types of submissions being accepted by the regulatory authority in the eCTD electronic-only filing format. Effective immediately, HC now also accepts two new submission types in the eCTD electronic-only filing format:
Notifiable Changes (NC)
Responses to Clarifaxes (If related to a submission filed in eCTD format).
Previously, HC accepted the eCTD electronic-only filing format for the following submission types only:
For More Information Please Click: Ask Cato - Health Canada Expands Acceptance of eCTD Submissions
GlobalSubmit Lunchtime Web Series

Join us for an upcoming web demonstration to discover the advantages of using GlobalSubmit 2010.
See how the FDA views electronic submissions
Build your application as the FDA sees it
Cut days off the time it takes to quality check bookmarks and hyperlinks
Improve your communications both internally and with the Agency
Improve your team’s efficiency and meet important deadlines
Reduce your risk of technical rejections
For Registration Please Click: GlobalSubmit Lunchtime Web Series
Subscribe to:
Posts (Atom)